SARUMAN
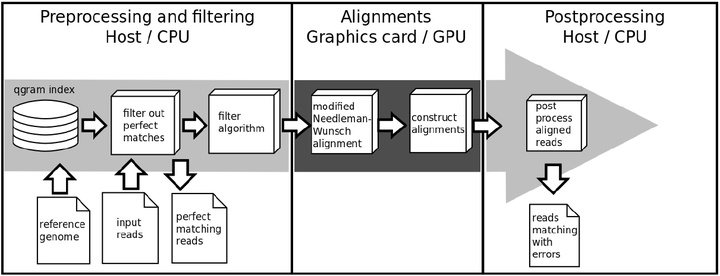
Using GPU programming for short read mapping.
Since the introduction of next generation sequencing technologies like Solexa, 454, and SOLiD the amount of generated data rises with each new technology upgrade. As the application scenarios especially of the short read techniques include the re-sequencing of known genomes or sequencing of closely related strains, new software tools are needed for the fast mapping of sequencing reads against a reference genome.
Currently, there are several tools available, but most of them are limited either in speed or accuracy. Limitations in accuracy lead to non detected mappings, which could become important in post processing steps like SNP calling. Because of those limitations our goal was to develop an exact and complete mapping algorithm with equivalent running time compared to available heuristic implementations.
The result is SARUMAN (Semiglobal Alignment of Short Reads using CUDA and Needleman-Wunsch). SARUMAN uses a qgram index based filter algorithm followed by a modified Needleman-Wunsch alignment. To speed up he normally time-consuming alignment step all alignments are processed on a NVIDIA graphics card to exploit the massive parallel architecture of new graphics processing units (GPUs). Based on this technique, depending on the input read length, SARUMAN is able to process hundreds of thousands of alignments in just a few seconds. As a result of this alignment strategy SARUMAN not only detects mismatches, but also allows to detect and handle all insertions and deletions correctly. The mapping algorithm is exact and complete, it identifies all possible matching positions for a given error threshold and always returns the optimal local alignment.